Highlights from SeraCare's AACR 2019 meeting experience
The team at SeraCare have been experiencing the sights and sounds of the annual AACR meeting this week in Atlanta, and they are exciting about what they're learning! Catherine Huang, Director of Product Development at SeraCare, shared her top highlights from Atlanta this week. See what she had to say here!
Day One:
I’m excited to be at the Annual AACR meeting at the Georgia World Congress Center in Atlanta, GA. AACR is my favorite scientific meeting because each year I am inspired by the remarkable research presented and leave the conference feeling I’ve learned an incredible amount in just a few days. This year, I thought I would recap some of the most exciting and incredible talks for those who weren’t able to make it to Atlanta.
The highlight for me today was the presentation by Crystal L. Mackall of Stanford University in the Opening Plenary Session titled “Next-generation CAR T cells designed to overcome tumor resistance.” This talk was so compelling because Dr. Mackall presented experiment data showing that more effective cellular therapies for cancer are possible.
CD19 CAR therapy (chimeric antigen receptor T cell therapy) was the first FDA-approved cell therapy: Kymriah® was approved for children with B-cell acute lymphoblastic leukemia in 2017. Approximately 40% of patients treated have sustained disease control. This is an amazingly successful therapy, but still only a minority of patients benefit. Dr. Mackall’s talk first focused on why the treatment is unsuccessful in the ~60% who don’t have durable responses.
One reason that the CD19 CAR T treatment fails is tumor heterogeneity, i.e. the tumor changes so that it loses the CD19 antigen that is recognized by the T cell. This is also known as “antigen loss escape.” There may be selection of tumor cells with variant splice isoforms that lack the exon to which the antibody binds. Therefore, we need to be able to target more than one antigen. Another pan-B cell antigen, CD-22, was also identified as an effective target for CAR T therapy. However, the same issue of antigen loss or downregulation arises when patients are on the CD-22 therapy. Even a modest downregulation of approximately two-fold can make CAR T cell killing less effective. By generating CAR T cells with bivalent specific receptors to CD19 or CD22, the CAR T therapy can target two antigens simultaneously and overcome the problem of antigen loss escape.
The second reason that CD19 CAR T therapy fails is due to intrinsic T cell dysfunction and T cell exhaustion. T cell exhaustion is correlated with poor cellular expansion and early loss of the CAR T cells within the patient. T cells may already be exhausted at the onset of therapy, or because the tumor burden is so high that the CAR T cells are rapidly exhausted by the constant signaling. PD-L1 checkpoint blockage can be helpful in relieving exhaustion but does not completely restore function.
Dr. Mackall’s lab has used an in vitro model for T cell exhaustion in which there are tonically signaling CARs which produce very little IL-2 and terminally differentiate. From this model, they have learned that T cell exhaustion is an epigenetic event. By using ATAC-seq studies, they looked at regions of the genome that are in open vs. closed transcriptional states in the exhausted state. They found that AP-1/bZIP/IRF transcription factors are over-expressed in exhausted T cells. They concluded that AP-1 transcription factors encode a transcriptional program that is inhibitory to T cell activation. Dr. Mackall went on to explain that while activation stimulatory factors like Fos and cJun may be present in exhausted CAR T cells, other potentially inhibitory AP-1 factors like JunB, BATF, ATF, and IRF are overexpressed to a greater degree, so it’s the imbalance of stimulatory and inhibitory AP-1 family members that may lead to exhaustion.
The real excitement of the talk came when she described how her lab has used c-Jun overexpression to restore the balance and reactivate exhausted T cells. C-Jun overexpression not only improves anti-tumor activity through IL-2 and interferon gamma cytokine production, but there is also less PD-L1 checkpoint expression. It appears to also help mitigate low antigen escape because the c-Jun-overexpressing cells can still activate killing even when the antigen is present at relatively low levels. These data provide hope that exhaustion-resistant CAR T cells may be possible, and may represent more effective therapy for patients.
We’re presenting our own exciting data on reference standards for TMB, liquid biopsy, NTRK, fusion RNA, and more at the AACR poster sessions. You can find a full listing here, and a form for free downloads.
Day Two:
At the AACR Annual Meeting, I was most excited to attend the major symposium entitled “The Microbiome as an Orchestrator of Immunity and Cancer Immunotherapy,” which featured three highly informative talks. First, Gregory Sonnenberg from Weill Cornell Medicine gave an overview of how the microbiome contributes to immune homeostasis in the intestine. Second, Carrie Daniel-MacDougall of MD Anderson Cancer Center talked about how diet and lifestyle affect the microbiome. The third and most exciting presentation, in my opinion, was given by Giorgio Trinchieri from the NCI, who talked about the microbiome in cancer therapy.
In Dr. Trinchieri’s presentation, he talked about how the progression of cancer is affected by the microbiome due to its major role in inflammation and immunity. However, the question he has set out to answer is how the composition of the gut microbiome can affect the efficacy of treatment. His findings indicate that the microbiome appears to affect therapy outcomes by “training” the myeloid and innate immune components at distant tumor sites; possibly priming myeloid cells to produce cytokines that may improve therapeutic responses.
Three separate clinical studies were recently published1-3 which indicate that the composition of the gut microbiome modulates the effects of anti-PD1 therapy. All three studies found that patients can be stratified into responders and nonresponders to immunotherapy based on the composition of their gut microbiomes. Interestingly, these studies, which were performed in different cities (Houston, Chicago, and Paris) using different cancer types, came up with different species of bacteria that were most correlated with response to immunotherapy. Why the discordant results? Dr. Trinchieri hypothesized that because environment dominates over host genetics in shaping the gut microbiome, perhaps the discordance was due to the populations being studied in various geographic locations.
Dr. Trinchieri posed that the goal of future research should be to precisely identify the bacteria species that favor anti-PD1 therapy response and characterize their molecular mechanisms. He noted that machine learning can be used for predicting patients’ anti-PD1 therapy response with 70-80% accuracy within one cohort, but when used to train for predicting outcomes for a different cohort, the accuracy is no better than flipping a coin. This means we need to discover microbiome biomarkers for prediction of response across geographies, which is currently lacking. Despite the infancy of our understanding of microbiome biomarkers, modulation of the microbiome can still have beneficial effects. Dr. Trinchieri told of a small pilot study at the University of Pittsburgh in which patients resistant to anti-PD1 therapy underwent fecal transplant from responders and then additional immune checkpoint therapy. So far, two out of three patients are showing some response to this treatment (stable disease or regression).
This talk, and the others in the session, indicated to me how the microbiome has catapulted into a central role in understanding immuno-oncology. As we’re developing and implementing cancer genotyping assays to predict response to I-O therapies (and corresponding reference standards), we need to consider these non-genomic factors that play a critical, but not-yet fully understood function. I can’t wait to hear what progress will be made in the coming year.
Have you seen our AACR 2019 posters? You can download our featured presentations on reference standards for TMB, liquid biopsy, NTRK, fusion RNA, and more by clicking here.
Day Three:
My third day at the AACR Annual meeting was a day of phenomenal presentations. I am struggling to choose just one to tell you about because I attended multiple inspiring, thought-provoking, and even entertaining talks today. I decided to report on the plenary presentation by Steven A. Rosenberg entitled “T-cell therapy targeting unique cancer mutations” because I think this story has the most potential to positively impact patient outcomes.
Dr. Rosenberg started his presentation by saying that solid epithelial cancers often don’t respond well to therapy. His team’s approach has been to use adoptive immunotherapy, which utilizes TILs (tumor-infiltrating lymphocytes) grown in vitroand then transferred back to the patient. The advantage of this method is that you can select with high affinity to cancer antigens and activate the T cells prior to transferring back to the patient. Additionally, you can manipulate the patient host prior to re-infusion by chemotherapy or lymphodepletion. Approximately 55% of patients achieve a durable response to this therapy.
Now Dr. Rosenberg’s research has focused on what the TILs actually recognize that enable the destruction of the tumor cells. To activate the TILs, Dr. Rosenberg explained that cancer antigens must be processed into an approximately 9-11 amino acid peptide that fits into the groove of an MHC molecule. So, only a very few cancer mutations will be immunogenic. His lab has surveyed the mutations present in a particular patient, then made all possible combinations of peptides with the mutation. Minigenes encoding these peptides are transfected into antigen-presenting cells (APCs), which are then co-cultured with the patient’s own T cells. They test to see if any of the peptides presented on the surface of the APCs can be recognized by the T cells. Dr. Rosenberg stressed that there was no prediction involved in this work; models generally don’t accurately predict tumor antigens. In this approach, each mutation is empirically tested.
The immunogenic mutations identified seemed to be unique to each patient. For example, when they looked at gastrointestinal cancers, they examined mutations from 72 patients and identified 120 immunogenic peptides; only one of which was shared among two patients – none of the mutations identified as immunogenic were driver mutations. This work led the Rosenberg group to pose two hypotheses: first, it is the recognition of random somatic mutations that is the final common pathway explaining cancer regression from immunotherapy, and second, any intracellular protein can potentially be a cancer antigen.
There is good news and bad news that comes out of these hypotheses: the good news is virtually all cancers have mutations that can be neoantigens; the bad news is the process to identify these neoantigens must be highly customized for each patient. Ongoing work is attempting to make the process less customized and therefore less labor intensive, so that the therapy can be offered to a greater number of patients. He noted that there are many mutations in driver oncogenes that can be shared among patients. For example, 51% of pancreatic cancer patients have a mutation at glycine 12 of KRAS. Making a KRAS G12 peptide library and preparing APCs for screening could be beneficial to a large portion of pancreatic cancer patients. P53 is a tumor suppressor gene that is also frequently mutated, and the mutations cluster in a limited number of “hotspots” in the gene. Preparing a peptide library for these hotspot regions of the protein could also potentially be a tool applicable for many patients.
The selected T cells that are re-infused into the patient can expand 1,000-fold in the days after transfer and persist at low levels indefinitely. Visuals from clinical cases are always very impactful for me, and Dr. Rosenberg showed several images of patients with bodies racked with metastatic lesions that were completely resolved after the adoptive T cell therapy. It was truly awe-inspiring to see how this innovative method was able to completely turn around the prognosis for the sickest patients and restore them to health. I am very hopeful that current work in the Rosenberg lab, and other labs, will continue to refine and make this therapy more widely available.
Don’t miss out on the latest data we presented at AACR on reference standards for TMB, liquid biopsy, NTRK, fusion RNA, and more in the poster sessions. You can download our posters here.
Day 4: Presenting NTRK Reference Materials for Global Assay Standardization at AACR 2019
On the last morning of AACR 2019, I had the privilege of presenting a poster together with my colleague, Sebastian Bender from Bayer AG, in Berlin. Because of this, I didn’t have a chance to attend any talks, but I still wanted to finish out my blog series with highlights from each day of the conference. The poster session was a personal highlight of the meeting because of all the great feedback we received on the project. I want to share with you a little about the poster we presented called “Development of NTRK Reference Materials for Global Assay Standardization,” which is available for you to download along with our other featured posters.
This poster came out of a collaboration between SeraCare, Bayer AG, and Bayer US that identification of genomic alterations underlying carcinogenesis through genomic testing is critical to match patients to either an approved cancer therapy or a drug in a clinical trial. Chromosomal rearrangements of the three neurotrophic tyrosine receptor kinase genes (NTRK1, NTRK2, NTRK3) that encode tropomyosin receptor kinases (TRK) are genomic alterations that drive oncogenesis in many adult and pediatric cancers. In TRK fusion cancer, the NTRK gene fuses with an unrelated gene, causing overexpression of the oncogenic TRK fusion protein.
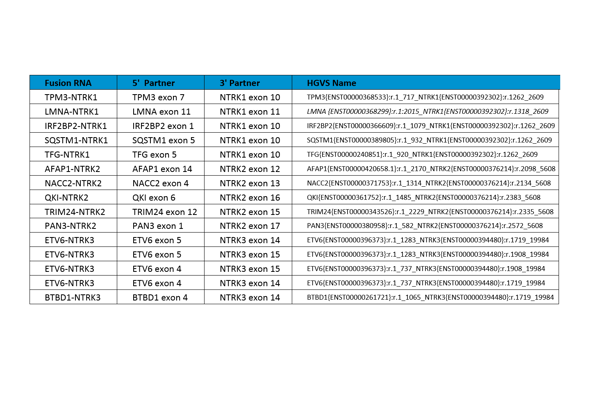
Accurate and reliable genomic testing for NTRK fusions has become critically important because of innovative precision biopharmaceutical treatments such as larotrectinib, which was approved by the US Food and Drug Administration (FDA) for the treatment of patients with locally advanced or metastatic solid tumors harboring NTRK gene fusions. In this study, we aimed to develop a 15-plex reference standard which can be used to evaluate genomic NTRK testing, optimize assays, tune bioinformatics pipelines, and benchmark among testing sites.
The criteria for selecting the 15 NTRK fusion RNAs in the reference standard included:
- Fusions involving all three NTRK genes (five fusions each for NTRK1, NTRK2, and NTRK3).
- Prevalence data for NTRK fusions published in three larotrectinib studies (Drilon et al. 2018; NEJM; Table S2) with all selected fusions present in at least two patients.
- Prevalence data for NTRK fusions in the COSMIC database (as of May 2018).
After the NTRK fusions were selected, DNA constructs containing the fusions of interest were synthesized, and biosynthetic fusion RNAs were transcribed in vitro. GM24385 cell line was modified to contain the biosynthetic NTRK RNA fusions. Cells were fixed in 10% buffered formalin for 18 hours, embedded in paraffin, and 10-micron curls were sectioned. Curls were extracted and tested using in-house developed digital PCR assays run on Bio-Rad Qx200™. Digital PCR is an absolute quantitation method and gives an accurate measure of the copies of each biosynthetic fusion RNA per nanogram of total RNA extractable from the curl.
Archer® FusionPlex® Solid Tumor Kit run on Illumina MiSeq® and OncomineTM Comprehensive Assay were used to demonstrate that the reference standard is compatible with commonly used commercial NGS assays. You can download the poster here to review the results.
There was a lot of discussion among poster attendees about why the percent of reads supporting the fusions varies so much on the Archer assay (6.2% for LMNA-NTRK1 up to 63.1% for AFAP1-NTRK2). The Archer analysis software defines this metric as “percent of reads supporting the event. It is the number of unique reads spanning the breakpoints and supporting the event, divided by the total number of unique RNA reads that span either breakpoint. Reads from all primers are combined for this calculation (but only those reads spanning the breakpoint on either side of the junction).”
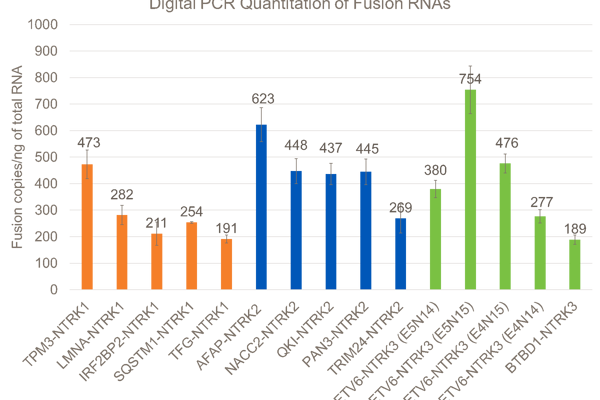
So, it appears that if both of the fusion partner transcripts are poorly expressed in the GM24385 reference cell lines, most of the reads will be from the biosynthetic fusion transcripts, and the percent of reads supporting the fusion will be high. However, if either of the partner transcripts are highly expressed in GM24385 (like LMNA is), then many reads will come from the endogenous transcripts and the percent of reads supporting the fusion will be low.
Another thing I learned from conversations at this poster is that labs are using a wide variety of assays beyond the Archer and Oncomine platforms -- some of which score for 5’ and 3’ imbalance of exon expression across key transcripts. I gained so much insight and knowledge in Atlanta, and I’m hoping to gather some more data for this product on diverse testing systems in our lab.
The key takeaways from our poster were that high-quality genomic cancer testing is essential for identifying targetable alterations in cancer patients who may benefit from precision therapies. Highly-characterized, patient-like reference standards such as the Seraseq® FFPE NTRK Fusion RNA Reference Material described here can be used to standardize testing across laboratories and clinical sites. This reference standard contains 15 clinically relevant NTRK fusions in a single reference sample and is compatible with different NGS testing methodologies.
This was one of several great posters that we and our collaborators presented at AACR on topics from TMB to liquid biopsy. You can download them all for free right here.
These highlights were originally published on the SeraCare blog.